
Vérification de sécurité / Security check / Verificación de seguridad
Par mesure de sécurité, nous avons besoin de vérifier que vous êtes bien un utilisateur humain.
Pour celà, nous vous demandons d'écrire le texte affiché dans l'image ci-dessous :
For security reasons, we need to verify that you are a human user.
To do this, please type the text shown in the image below:
Por motivos de seguridad, necesitamos comprobar que eres un usuario humano.
Para ello, te pedimos que escribas el texto que aparece en la imagen siguiente :
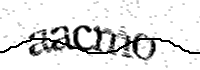
Référence / Reference / Referencia : 300